Cytogenetic, Teratogenic, and Miscellaneous Other Disorders Causing Male Pseudohermaphroditism or Germ-Cell Failure in Males (46,XY) and Females (46,XX)
Authors
INTRODUCTION
In male pseudohermaphroditism, individuals with a Y chromosome have external genitalia that fail to develop as expected for normal males. In 46,XY disorders of sex development (DSD) external genitalia are sufficiently ambiguous to confuse the gender of rearing. Male pseudohermaphroditism encompasses a wide spectrum of disorders as shown in Table 1. Many of these disorders have been discussed in previous chapters. In this chapter teratogenic, cytogenetic (45,X/46,XY) and genetic forms of male pseudohermaphroditism not reviewed elsewhere are discussed.
Table 1. The spectrum of 46,XY gender reversal (XY females)
XY gonadal dysgenesis without somatic anomalies
Perturbations of SRY (HMG-box)
Duplication Xp (DAX1)
X-linked recessive form
Forms without detectable molecular perturbation or heritability
XY gonadal dysgenesis and Wilms’ tumor oncogene (WT1)
Denys-Drash syndrome
Frasier syndrome
XY gonadal dysgenesis with malformation
XY gonadal dysgenesis and campomelic dysplasia (SOX9)
XY gonadal dysgenesis and α-thalassemia X chromosome (ATX) syndrome
XY gonadal dysgenesis in other malformation syndromes
Ectodermal anomalies (Brosnan)
Genital-palato-cardiac (Gardner-Silengo-Wachtel)
Spastic paraplegia-optic atrophy-microcephaly (Teebi)
XY gonadal dysgenesis with autosomal deletions
Del (2p)
Del (9p)
Del (10q)
XY gonadal dysgenesis with autosomal duplications
Dup1p (WNT1)
Dup17q (SOX9)
Germ-cell failure in both sexes (46,XY cases)
No somatic anomalies
Hypertension and deafness
Alopecia
Microcephaly and short stature
Leydig cell hypoplasia
Steroid biosynthetic defects
Steroidogenic factor 1 deficiency (StAR)
17α-hydroxylase/17,20-desmolase deficiency (CYP17)
3β-ol dehydrogenase/3β-hydroxysteroid dehydrogenase deficiency (3βHSD)
Agonadia (46,XY)
TERATOGENIC MALE PSEUDOHERMAPHRODITES
Despite many claims of genital abnormalities being caused by putative teratogens, virtually no proven examples exist. Nonetheless, if administered in sufficiently high doses in the first trimester to a woman pregnant with a male fetus, several agents would be expected to produce female external genitalia. One example is cyproterone, the mode of action of which is to block the androgen receptor. Cyproterone is present in oral contraceptive formulations in Europe, but not in the United States. Little reported information exists on the consequences of inadvertent ingestion during pregnancy, but presumably problems have either not occurred or not been reported. On the other hand, genital ambiguity was unexpectedly reported in association with trimethadione teratogenicity.1 If valid, the mechanism by which this occurred is unclear.
Another potential teratogen is finasteride, which inhibits 5α-reductase. Perturbing this function (5α-reductase) causes male pseudohermaphroditism (see Chapter 5.80). Another agent is flutamide. Both finasteride and flutamide are approved by the US Food and Drug Administration (FDA) for treatment of hirsutism; thus, the risk for teratogenic male pseudohermaphroditism exists. Again, no cases of human teratogenicity appear to have been claimed to be associated with these agents.
Controversy exists concerning whether administration of progestins or progesterones during pregnancy can produce simple hypospadias, that is, without genital ambiguity per se. The overwhelming weight of the evidence seems to be that these agents do not adversely affect male genital development.2, 3
In general, C-21 progestogen derivatives (e.g., medroxyprogesterone) do not virilize even in high doses; 19-nortestosterone derivatives (e.g., norethindrone) generally virilize, but there are exceptions (e.g., norethynodrel). In 1962, Jacobson4 reported an 18% incidence of masculinization in female infants whose mothers were given high doses of norethindrone acetate for pregnancy maintenance; virilization occurred in only 1% exposed to medroxyprogesterone acetate. Doses of 19-nortestosterones required for virilization are 10–20 mg/day, far in excess of that associated with inadvertent contraceptive exposure during pregnancy. When progestogen therapy at such dosage is currently administered, the compound administered will almost certainly be progesterone, 17α-hydroxyprogesterone caproate, or medroxyprogesterone. Genital ambiguity as a result of progestogen exposure of female fetuses is thus mostly a topic of historic concern.
The only currently utilized sex steroid that causes virilization when administered in usually administered doses is danazol, a derivative of 17α-ethinyl testosterone. In the study by Brunskill5 on adverse drug reports of pregnant women receiving danazol, 23 of 57 female infants were virilized; male offspring were ostensibly normal. Genital virilization has resulted from doses as low as 200 mg daily, whereas 800 mg daily is the usual dose when danazol is used to treat endometriosis.
CYTOGENETIC MALE PSEUDOHERMAPHRODITISM
45,X/46,XY individuals have a 45,X cell line and at least one cell line containing a Y chromosome. Based on cohort studies of 45,X/46,XY cases detected in utero (prenatal genetic diagnosis), more than 90% of cases are normal males.6 The phenotype of cases ascertained postnatally differs markedly, doubtless reflecting selection basis. Postnatal cases manifest a variety of phenotypes but predominately genital ambiguity or phenotypically normal females.7, 8, 9 Different phenotypes presumably reflect different tissue distributions of the various cell lines.
Of note, structurally abnormal Y chromosomes are not infrequently present. Given that structurally abnormal chromosomes (e.g., dicentric) are often unstable, it is likely that the 45,X line arises secondarily after loss of the structurally abnormal Y. This may explain why certain infertile males with normal external genitalia, for example requiring intracytoplasmic sperm injection (ICSI), may have offspring with genital ambiguity. In the sons a 45,X line might have arisen secondarily.
45,X/46,XY with unambiguous female external genitalia
These 45,X/46,XY individuals may have Turner’s stigmata and be clinically indistinguishable from 45,X individuals in many ways. However, 45,X/46,XY cases may be normal in stature and show no somatic anomalies. External genitalia, vagina, and Müllerian derivatives remain unstimulated because of the lack of sex steroids. Breasts fail to develop, and little pubic or axillary hair develops. If breast development occurs in a 45,X/46,XY individual, an estrogen-secreting tumor such as gonadoblastoma or dysgerminoma should be suspected.10 However, virilization has also been claimed to result from gonadotropin stimulation of streak gonads;11 studies of such cases would be welcome.
Although streak gonads of 45,X/46,XY individuals may be histologically indistinguishable from streak gonads of 45,X individuals, gonadoblastomas or dysgerminomas develop in approximately 15–20% of 45,X/46,XY individuals.12 If retained in a phenotypic XY female, presence of the Yq locus GBY (gonadoblastoma Y chromosome) is believed to predispose to neoplasia. If GBY is deleted, as it is in many deletions of Yq, risk of neoplasia is diminished in gender-reversal females.13
Neoplasia may develop in the first or second decade of life. Even if the presumptive GBY locus were to be absent, we recommend gonadal extirpation for all 45,X/46,XY individuals having female external genitalia. The uterus should be retained, given that pregnancy may be achievable through donor oocytes or donor embryos.
Gonadectomy can usually be accomplished by laparoscopy14 (Fig. 1). Although preferable to removing only gonads, adnexal removal may technically be necessary as well. Only rarely should laparotomy prove necessary.
|
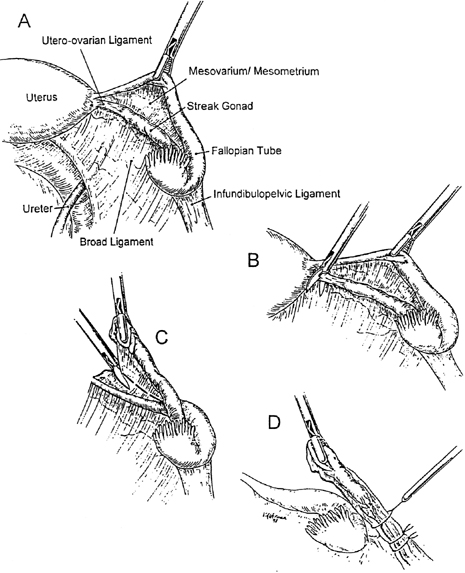
45,X/46,XY with ambiguous external genitalia
The term asymmetric or mixed gonadal dysgenesis applies to individuals with one streak gonad and one dysgenetic testis. These individuals usually have ambiguous external genitalia and a 45,X/46,XY complement, and usually a uterus. Many investigators believe that the phenotype is invariably associated with 45,X/46,XY mosaicism. Occasionally only 45,X or only 46,XY cells are demonstrable. Ostensibly, nonmosaic cases may merely reflect inability to sample sufficient tissues or cells.
45,X/46,XY individuals with ambiguous external genitalia usually have Müllerian derivatives (e.g., a uterus). Presence of a uterus is important diagnostically because that organ is absent in almost all genetic (Mendelian) forms of male pseudohermaphroditism. If an individual shows ambiguous external genitalia, bilateral testes, and a uterus, it is reasonable to infer 45,X/46,XY mosaicism actually exists. This statement still applies if both lines cannot be demonstrated cytogenetically. Occasionally the uterus may be rudimentary, or a fallopian tube may fail to develop ipsilateral to a testis.
45,X/46,XY with nearly normal male external genitalia
45,X/46,XY mosaicism may be detected in individuals with nearly normal male external genitalia. In fact, this is the most common 45,X/46,XY phenotype based on liveborn follow-up of 45,X/46,XY fetuses ascertained at amniocentesis, 90% of whom show a normal male phenotype.15 That 45,X/46,XY neonates far more commonly show genital ambiguity merely reflects biases of ascertainment. Those with male phenotype pass unrecognized.
45,X/46,XY individuals having almost normal male external genitalia do not seem to develop neoplasia as often as 45,X/46,XY individuals having female or frankly ambiguous genitalia.16 If gonads can be assessed periodically within the scrotum by ultrasound or palpation, gonadal extirpation should not be necessary if the male gender of rearing is chosen.
MALE PSEUDOHERMAPHRODITISM ASSOCIATED WITH MULTIPLE MALFORMATION SYNDROMES
Genital ambiguity may occur in 46,XY individuals having various multiple malformation syndromes. These include the Meckel-Gruber syndrome, Smith-Lemli-Opitz syndrome, brachio-skeletal-genital syndrome,17 esophageal-facial-genital syndrome,18 and other disorders. These disorders are usually inherited in either autosomal recessive or X-linked recessive fashion, and are summarized in Table 1. Many additional syndromes are associated with cryptorchiditism or simple hypospadias, but these rarely pose diagnostic problems.19 Of relevance as well are existence of various syndromes associated with XY gender reversal. These are listed in Table 2 and are discussed in Chapter 5.87. These syndromes include campomelic dysplasia (SOX-9), Denys-Drash and Frasier syndromes (WT-1) autosomal deletions (1q, 2q, 9p, 10q), and autosomal duplication (1q). In each condition the phenotype of affected 46,XY individuals is usually complete gender reversal (female phenotype). However, varied expression exists, some affected cases may occasionally present as male pseudohermaphrodites (genital ambiguity).
Table 2. Multiple malformation syndromes associated with ambiguous genitalia
Syndrome | Prominent features | Etiology |
Ablepharon-macrostomia20 | Absent eyelids, eyebrows, eyelashes, external ears; fusion defects of the mouth; ambiguous genitalia; absent or rudimentary nipples; parchment skin, delayed development of expressive language | Autosomal recessive |
Moderate to severe mental deficiency, growth deficiency, microcephaly, aniridia, nystagmus, ptosis, blindness, Wilms' tumor, ambiguous genitalia, gonadoblastoma | Chromosomal or autosomal dominant (WT-1) | |
Asplenia, cardiovascular anomalies, and caudal deficiency23 | Hypoplasia or aplasia of the spleen, complex cardiac malformations, abnormal lung lobulation, anomalous position and development of the abdominal organs, agenesis of corpus callosum, imperforate anus, ambiguous genitalia, contractures of the lower limb | Autosomal recessive |
Beemer24 | Hydrocephalus, dense bones, cardiac malformation, bulbous nose, broad nasal bridge, ambiguous genitalia | Autosomal recessive |
Deletion 11q25 | Trigonocephaly, flat and broad nasal bridge, micrognathia, carp mouth, hypertelorism, low-set ears, severe congenital heart disease, anomalies of limbs, external genitalia | Chromosomal |
Wilms' tumor, nephropathy, ambiguous genitalia with 46,XY karyotype | Unknown | |
Cryptophthalmia, defect of auricle, hair growth on lateral forehead to lateral eyebrow, hypoplastic nares, mental deficiency, partial cutaneous syndactly, urogenital malformation | Autosomal recessive | |
Lethal acrodysgenital dysplasia30 | Failure to thrive, facial dysmorphism, ambiguous, genitalia, syndactyly, postaxial polydactyly, Hirschprung disease, cardiac and renal malformations | Autosomal recessive |
Rutledge31 | Joint contractures, cerebellar hypoplasia, renal hypoplasia, ambiguous genitalia, urologic anomalies, tongue cysts, shortness of limbs, eye abnormalities, heart defects, gallbladder agenesis, ear malformations | Autosomal recessive |
SCARF syndrome32 | Skeletal abnormalities, cutis laxa, craniosynostosis, ambiguous genitalia, psychomotor retardation, facial abnormalities | Uncertain |
Short stature; short limbs; cleft lip and palate; ear anomalies; limb anomalies, including pre- and postaxial polysyndactyly; narrow thorax; short horizontal ribs; high clavicles; ambiguous genitalia | Autosomal recessive | |
Microcephaly, mental retardation, hypotonia, ambiguous genitalia, and sometimes gender-reversed abnormal facies | Autosomal recessive (deficiency 7-OH cholesterol dehydrogenase) | |
Trimethadione, teratogeneicity1 | Mental deficiency, speech disorders, prenatal onset growth deficiency, brachycephaly, midfacial hypoplasia, broad and upturned nose, prominent forehead, eye anomalies, cleft lip and palate, cardiac defects, ambiguous genitalia | Teratogenicity |
Vertebral, anal, tracheoesophageal, and renal anomalies; subjects with ambiguous genitalia as part of the cloacal anomalies | Unknown (if valid entity); alleged progestational teratogeneicity unproved (see Simpson and Kaufman2) | |
Genital–Palato-Cardiac syndrome (Gardner- Silengo-Wachtel)39 | Cleft palate, micrognathia, low-set ears, ventricular septal defect, internal anomalies; female to male external genitalia with varied gonads (streaks to testes) | X-linked or autosomal recessive |
(Simpson JL, Elias S: Genetics in Obstetrics and Gynecology. Philadelphia, WB Saunders, 2003.)
Two multiple malformation syndromes characterized by genital ambiguity (i.e., Smith-Lemli-Opitz syndrome and genito-palato-cardiac [Gardner-Silengo-Wachtel] syndrome) are of special interest to obstetrician–gynecologists.
SMITH-LEMLI-OPITZ SYNDROME
In this common autosomal recessive syndrome, 46,XY individuals show genital abnormalities ranging from hypospadias to genitalia ambiguity. In Ontario, the incidence has been estimated at 1 per 22,700 among individuals of European ancestry.40 An estimate of 1 per 20,000 is accepted.41 A characteristic spectrum of dysmorphic features allows diagnosis by experienced clinicians and geneticists. Mental retardation exists as does craniofacial dysmorphia characterized by microcephaly, low-set ears, ptosis, anteverted nares, inner epicanthal folds, broad maxillary ridges, and micrognathia. Syndactly of the third toe is present. Ureteropelvic junction obstruction and renal anomalies (cystic dysplasia, agenesis, duplication of kidneys) are also common. Approximately two-thirds have genital problems: usually hypospadias, micropenis, or hypoplastic scrotum are the manifestations; genital ambiguity is less common.
However, a more severe phenotype of Smith-Lemli-Opitz syndrome exists, often called type II. Here external genitalia may be female (gender reversal).42 Both type I and type II Smith-Lemli-Opitz syndrome, if the distinction is truly appropriate, are caused by mutation of a gene the product of which is C7-reductase, the enzyme responsible for converting 7-hydroxycholesteral to cholesterol.43, 44 A defect in exon–intronic splicing is the most common molecular perturbation.
Considerable attention is being directed to the feasibility of postnatal as well as prenatal treatment with a high-cholesterol diet. Given its low molecular weight, cholesterol should cross the placenta, making this mode of therapy feasible. During pregnancy, maternal serum estriol is low to nondetectable,45 making detection possible during maternal serum analyte screening. Maternal serum analyte screening is based on low maternal serum alpha-fetoprotein (MSAFP; multiple of median [MOM] 0.72), μE3 (MOM 0.21), and human chorionic gonadotropin (hCG; MOM 0.76).41 An algorithm based on performing an invasive procedure when risk is 1:100 will yield 71% detection at a procedure rate of only 0.44%.41 Definitive prenatal diagnosis is facile with amniotic fluid, based on molecular studies or presence of the novel compounds dehydroestriol and dehydropregnanetriol. Diagnosis can even be made in maternal urine, as in normal pregnancies these compounds are undetectable.46
AGONADIA
In agonadia, the gonads are completely absent, not merely present in the form of streaks as in gonadal dysgenesis. In agonadia the external genitalia are abnormal but female-like. In the original cases of Overzier and Linden47 the clitoris was enlarged and a perineal urethral orifice present. The distance between urethral orifice and anus seemed normal despite nearly complete labio-scrotal fusion. In other cases external genitalia were more normally female in appearance. No more than rudimentary Müllerian or Wolffian derivatives are present.
In approximately half of the reported cases, somatic anomalies coexist. In considering their own and previous cases (Overzier and Linden,47 Chaptal and colleagues,48 Overzier,49 Schoen and colleagues50) in 1973 Sarto and Opitz51 wondered whether a distinctive somatic malformation pattern existed: skull abnormalities (turricephaly and turribrachycephaly); facial dysmorphia (ptosis, epicanthal folds, nystamus, esotropia); vertebral anomalies; and low posterior hairline.
Agonadia was once believed to be confined to 46,XY individuals. However, sporadic cases have been reported in 46,XX individuals.52, 53 In these sporadic 46,XX cases, there were no somatic anomalies, Mendonca and colleagues54 also reported agonadia without somatic anomalies in phenotypic sibs having unlike chromosomal complements (46,XY and 46,XX). Presumably the cases reported by Kennerknecht and colleagues55 represent a distinct syndrome: agonadism, hypoplasia of the pulmonary artery and lung, and dextrocardia in XX and XY sibs.
Pathogenic explanations initially focused on loss of testes early in embryogenesis, a logical hypothesis given that initially all cases were 46,XY. Even in these cases, however, not only absence of gonads, but also abnormal external genitalia and lack of internal genital ducts need to be considered. An attractive hypothesis is transient presence of fetal testes, sufficiently long to initiate male differentiation and suppress Müllerian differentiation but not long enough to complete male differentiation. Thus, the term testicular regression is favored by some. On the other hand, mechanisms not confined to testes must be invoked in 46,XX cases. Given both heritable tendencies51, 56 as well as somatic anomalies, a defect in connective tissue is a logical explanation.
Few molecular clues exist. No genetic perturbations have been reported in SRY.57, 58, 59 One heterozygous mutation was found in SF-1.60 Agonadia has also been observed in the CHARGE association (a polymalformative disease associating coloboma, heart disease, atresia of choanae, retarded growth and development, genital hypoplasia and ear anomalies).61
REFERENCES
Feldman GL, Weaver DD, Lovrien EW: The fetal trimethadione syndrome: Report of an additional family and further delineation of this syndrome. Am J Dis Child 131: 1389, 1977 |
|
Simpson JL, Kaufman R: Fetal effects of progestogens and diethylstilbestrol. In Fraser IS, Jansen RPS, Lobo RA, et al (eds): Estrogens and Progestogens in Clinical Practice. pp. 533, 553 London, Churchill Livingstone, 1998 |
|
Phillips OP, Simpson JL: Contraception and congenital malformations. In Sciarra JJ (ed): Gynecology and Obstetrics. pp. 1, 21 Philadelphia, Lippincott Williams and Wilkins, 2001 |
|
Jacobson RB: Hazards of norethindrone therapy during pregnancy. Am J Obstet Gynecol 84: 962, 1962 |
|
Brunskill PJ: The effects of fetal exposure to danazol. Br J Obstet Gynaecol 99: 212, 1992 |
|
Chang HJ, Clark RD, Bachman H: The phenotype of 45,X/46,XY mosaicism: An analysis of 92 prenatally diagnosed cases. Am J Hum Genet 46: 156, 1990 |
|
Simpson JL: Disorders of Sexual Differentiation: Etiology and Clinical Delineation. New York, Academic Press, 1976 |
|
McDonough PG, Tho SP: The spectrum of 45X/46,XY gonadal dysgenesis and its implications (a study of 19 patients). Pediatr Adolesc Gynecol 1: 1, 1983 |
|
Rosenberg C, Frota-Pessoa O, Vianna-Morgante AM, et al: Phenotypic spectrum of 45,X/46,XY individuals. Am J Med Genet 27: 553, 1987 |
|
Verp MS, Simpson JL: Abnormal sexual differentiation and neoplasia. Cancer Genet Cytogenet 25: 191, 1987 |
|
Bosze P, Szamel I, Molnar F, et al: Nonneoplastic gonadal testosterone secretion as a cause of vaginal cell maturation in streak gonad syndrome. Gynecol Obstet Invest 22: 153, 1986 |
|
Simpson JL, Photopulos G: The relationship of neoplasia to disorders of abnormal sexual differentiation. Birth Defects Orig Artic Ser 12(1): 15, 1976 |
|
Lukusa T, Fryns JP, van den BH: Gonadoblastoma and Y-chromosome fluorescence. Clin Genet 29: 311, 1986 |
|
Pisarska MD, Simpson JL, Zepeda DE, et al: Laparoscopic removal of streak gonads in 46,XY or 45,X/46,XY gonadal dysgenesis. J Gynecol Tech 4: 95, 1998 |
|
Hsu LY: Prenatal diagnosis of chromosome abnormalities through amniocentesis. In Milunsky A (ed): Genetic Disorders and the Fetus. pp. 155, Vol. 3: Baltimore, Johns Hopkins Press, 1986 |
|
Simpson JL, Photopulos G: Hereditary aspects of ovarian and testicular neoplasia. Birth Defects Orig Artic Ser 12(6): 51, 1976 |
|
el Sahy NI, Waters WR: The branchio-skeleto-genital syndrome. A new hereditary syndrome. Plast Reconstr Surg 48: 542, 1971 |
|
Opitz JM, Howe JJ: The Meckel syndrome (dysencephalic splanchnocystica, the Gruber syndrome). Birth Defects Orig Artic Ser 5: 167, 1969 |
|
Pinsky L, Beitel LK, Kazemi-Esfarjani P: Lessons from androgen receptor gene mutations that cause androgen resistance in humans. In LA Hughes (ed): Sex Differentiation: Clinical and Biological Aspects. pp. 95, England, Cambridge Serono Symposia Series [Frontiers in Endocrinology], 1996 |
|
Hornblass A, Reifler DM: Ablepharon macrostomia syndrome. Am J Ophthalmol 99: 552, 1985 |
|
Riccardi VM, Borges W: Aniridia, cataracts, and Wilms tumor. Am J Ophthalmol 86: 577, 1978 |
|
Riccardi VM, Sujansky E, Smith AC, et al: Chromosomal imbalance in the Aniridia-Wilms’ tumor association: 11p interstitial deletion. Pediatrics 61: 604, 1978 |
|
Rodriguez JI, Palacios J, Omenaca F, et al: Polyasplenia, caudal deficiency, and agenesis of the corpus callosum. Am J Med Genet 38: 99, 1991 |
|
Beemer FA, von Ertbruggen I: Peculiar facial appearance, hydrocephalus, double-outlet right ventricle, genital anomalies and dense bones with lethal outcome. Am J Med Genet 19: 391, 1984 |
|
Sirota L, Shabtai F, Landman I, et al: New anomalies found in the 11q-syndrome. Clin Genet 26: 569, 1984 |
|
Habib R, Loirat C, Gubler MC, et al: The nephropathy associated with male pseudohermaphroditism and Wilms’ tumor (Drash syndrome): A distinctive glomerular lesion. Report of 10 cases Clin Nephrol 24: 269, 1985 |
|
Turleau C, Niaudet P, Sultan C, et al: Partial androgen receptor deficiency and mixed gonadal dysgenesis in Drash syndrome. Hum Genet 75: 81, 1987 |
|
Greenberg F, Keenan B, De Yanis V, et al: Gonadal dysgenesis and gonadoblastoma in situ in a female with Fraser (cryptophthalmos) syndrome. J Pediatr 108: 952, 1986 |
|
Gattuso J, Patton MA, Baraitser M: The clinical spectrum of the Fraser syndrome: report of three new cases and review. J Med Genet 24: 549, 1987 |
|
Merrer ML, Briard ML, Girard S, et al: Lethal acrodysgenital dwarfism: A severe lethal condition resembling Smith-Lemli-Opitz syndrome. J Med Genet 25: 88, 1988 |
|
Rutledge JC, Friedman JM, Harrod MJ, et al: A “new” lethal multiple congenital anomaly syndrome: Joint contractures, cerebellar hypoplasia, renal hypoplasia, urogenital anomalies, tongue cysts, shortness of limbs, eye abnormalities, defects of the heart, gallbladder agenesis, and ear malformations. Am J Med Genet 19: 255, 1984 |
|
Koppe R, Kaplan P, Hunter A, et al: Ambiguous genitalia associated with skeletal abnormalities, cutis laxa, craniostenosis, psychomotor retardation, and facial abnormalities (SCARF syndrome). Am J Med Genet 34: 305, 1989 |
|
Cooper CP, Hall CM: Lethal short-rib polydactyly syndrome of the Majewski type: A report of three cases. Radiology 144: 513, 1982 |
|
Silengo MC, Bell GL, Biagioli M, et al: Oro-facial-digital syndrome II. Transitional type between the Mohr and the Majewski syndromes: report of two new cases Clin Genet 31: 331, 1987 |
|
Bialer MG, Penchaszadeh VB, Kahn E, et al: Female external genitalia and mullerian duct derivatives in a 46,XY infant with the Smith-Lemli-Opitz syndrome. Am J Med Genet 28: 723, 1987 |
|
Joseph DB, Uehling DT, Gilbert E, et al: Genitourinary abnormalities associated with the Smith-Lemli-Opitz syndrome. J Urol 137: 719, 1987 |
|
Sofatzis JA, Alexacos L, Skouteli HN, et al: Malformed female genitalia in newborns with the VATER association. Acta Paediatr Scand 72: 923, 1983 |
|
Kallen K, Mastroiacovo P, Castilla EE, et al: VATER non-random association of congenital malformations: Study based on data from four malformation registers. Am J Med Genet 101: 26, 2001 |
|
Greenberg F, Gresik MV, Carpenter RJ, et al: The Gardner-Silengo-Wachtel or genito-palato-cardiac syndrome: Male pseudohermaphroditism with micrognathia, cleft palate, and conotruncal cardiac defect. Am J Med Genet 26: 59, 1987 |
|
Nowaczyk MJ, McCaughey D, Whelan DT, et al: Incidence of Smith-Lemli-Opitz syndrome in Ontario, Canada. Am J Med Genet 102: 18, 2001 |
|
Palomaki GE, Bradley LA, Knight GJ, et al: Assigning risk for Smith-Lemli-Opitz syndrome as part of 2nd trimester screening for Down’s syndrome. J Med Screen 9: 43, 2002 |
|
Curry CJ, Carey JC, Holland JS, et al: Smith-Lemli-Opitz syndrome-type II: multiple congenital anomalies with male pseudohermaphroditism and frequent early lethality. Am J Med Genet 26: 45, 1987 |
|
Tint GS, Irons M, Elias ER, et al: Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N Engl J Med 330: 107, 1994 |
|
Tint GS, Batta AK, Xu G, et al: The Smith-Lemli-Opitz syndrome: A potentially fatal birth defect caused by a block in the last enzymatic step in cholesterol biosynthesis. Subcell Biochem 28: 117, 1997 |
|
Bradley LA, Palomaki GE, Knight GJ, et al: Levels of unconjugated estriol and other maternal serum markers in pregnancies with Smith-Lemli-Opitz (RSH) syndrome fetuses. Am J Med Genet 82: 355, 1999 |
|
Shackleton CH, Roitman E, Kratz L, et al: Dehydro-oestriol and dehydropregnanetriol are candidate analytes for prenatal diagnosis of Smith-Lemli-Opitz syndrome. Prenat Diagn 21: 207, 2001 |
|
Overzier C, Linden H: Echter agonadismus (Anorchismus) bei Geschwistern. Linden H Gynaecologia 142: 215, 1956 |
|
Chaptal J, Jean R, Pages R, et al: Sur un cas de dysgenesie gonadique avec manifestations androgeniques. Arch Fr Pediatr 15: 613, 1958 |
|
Overzier C: Echter agonadismus. In C Overzier (ed): Intersexualitas. pp. 348, 352 Stuttgart, Georg Thieme, 1961 |
|
Schoen EJ, King AL, Baritell A, et al: Pseudohermaphroditism with multiple congenital anomalies. Report of a case Pediatrics 16: 363, 1955 |
|
Sarto GE, Opitz JM: The XY gonadal agenesis syndrome. J Med Genet 10: 288, 1973 |
|
Duck SC, Sekhon GS, Wilbois R, et al: Pseudohermaphroditis, with testes and a 46, XX karyotype. J Pediatr 87: 58, 1975 |
|
Levinson G, Zarate A, Guzman-Toledano R, et al: An XX female with sexual infantilism, absent gonads, and lack of Mullerian ducts. J Med Genet 13: 68, 1976 |
|
Mendonca BB, Barbosa AS, Arnhold IJ, et al: Gonadal agenesis in XX and XY sisters: Evidence for the involvement of an autosomal gene. Am J Med Genet 52: 39, 1994 |
|
Kennerknecht I, Sorgo W, Oberhoffer R, et al: Familial occurrence of agonadism and multiple internal malformations in phenotypically normal girls with 46,XY and 46,XX karyotypes, respectively: A new autosomal recessive syndrome. Am J Med Genet 47: 1166, 1993 |
|
de Grouchy J, Gompel A, Salomon-Bernard Y, et al: Embryonic testicular regression syndrome and severe mental retardation in sibs. Ann Genet 28: 154, 1985 |
|
Pivnick EK, Wachtel S, Woods D, et al: Mutations in the conserved domain of SRY are uncommon in XY gonadal dysgenesis. Hum Genet 90: 308, 1992 |
|
Mendonca BB, Russell AJ, Vasconcelos-Leite M, et al: Mutation in 3 beta-hydroxysteroid dehydrogenase type II associated with pseudohermaphroditism in males and premature pubarche or cryptic expression in females. J Mol Endocrinol 12: 119, 1994 |
|
Zenteno JC, Jimenez AL, Canto P, et al: Clinical expression and SRY gene analysis in XY subjects lacking gonadal tissue. Am J Med Genet 99: 244, 2001 |
|
Philibert P, Zenaty D, Lin L et al: Mutational analysis of steroidogenic factor 1 (NR5a1) in 24 boys with bilateralanorchia: a French collaborative study. Hum Reprod. 2007 Dec;22(12):3255-61. Epub 2007 Oct 16. |
|
Kushnick T, Wiley JE, Palmer SM: Agonadism in a 46,XY patient with CHARGE association. Am J Med Genet. 1992 Jan 1;42(1):96-9. |